Caso clinico
Giugno 2017. Uomo di 71 anni, in vacanza sul lago di Garda, giungeva nel PS del nostro nosocomio per dispnea per sforzi di minima entità (classe funzionale NYHA III/IV); riferiva inoltre ortopnea e dispnea parossistica notturna, contrazione della diuresi e aumento ponderale.
Il paziente era seguito presso altro centro e dalla documentazione clinica portata in visione (ultimo controllo clinico circa 8 mesi prima) risultava essere affetto da:
- Cardiomiopatia ipertrofica con severa disfunzione diastolica e funzione sistolica ventricolare sinistra preservata (FE 55%)
- Insufficienza renale cronica II/III° stadio (GFR 40 ml/min – MDRD)
- Diabete mellito di tipo II
- Iperuricemia
- Ipercolesterolemia
Inoltre al controllo coronarografico del 2012 le coronarie risultavano essere indenni e nello stesso anno era stato sottoposto a impianto di PM bicamerale per malattia del nodo del seno con successivo up-grading del device ad ICD biventricolare nel 2014. Qualche anno prima riferiva inoltre di essere stato sottoposto a due interventi per sindrome del tunnel carpale bilaterale.
La terapia che il paziente stava assumendo era la seguente: Furosemide 175 mg/die; Irbesartan 75 mg/die; Atorvastatina 10 mg/die; Febuxostat 80 mg/die; Pantoprazolo 40 mg/die; Glimepiride 4 mg/die.
Obiettivamente erano presenti: un soffio sistolico 2/6 puntale e mesocardico; un murmure polmonare ridotto alla base destra, fegato palpabile a 2-3 dita dall’arcata costale, turgore giugulare e reflusso epato-giugulare positivo; edemi declivi improntabili bilaterali. Gli esami ematochimici mostravano lieve incremento della creatinina /1.57 mg/dL con ClCr 46 mL/min) e delle transaminasi (AST 67 U/L; ALT 88 U/L).
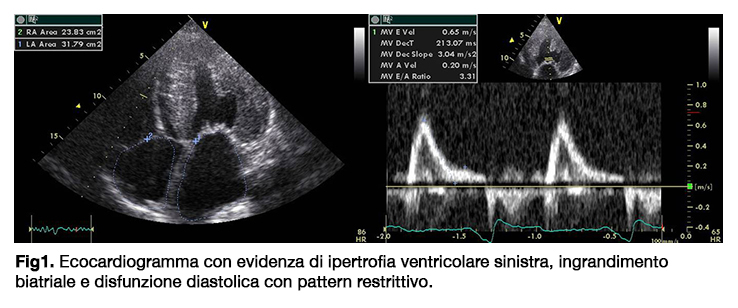
L’ecocardiogramma evidenziava severa ipertrofia ventricolare sinistra e funzione sistolica globale moderatamente ridotta (FE circa 40%); ventricolo destro anch’esso ispessito (13 mm) e ipocinetico (TAPSE 12; S’TVI 7 cm/sec); pattern di riempimento diastolico di tipo restrittivo (E >> A); dilatazione bi-atriale (area Asx 32 cm2); miitrale con lembi inspessiti, fibrotici con insufficienza valvolare degenerativa di grado moederato (2+/4+); IT moderata, stima PAPs 45-50 mmHg (se PVC = 15-20 mmHg) (Fig. 1)
 La radiografia del torace documentava la presenza di versamento pleurico basale destro di entità non elevata; accentuazione del disegno vascolare con ili polmonari un poco ampliati
La radiografia del torace documentava la presenza di versamento pleurico basale destro di entità non elevata; accentuazione del disegno vascolare con ili polmonari un poco ampliati
Il paziente è stato pertanto inizialmente trattato con terapia diuretica ev con progressivo miglioramento del quadro clinico e del compenso emodinamico (buona risposta diuretica e calo ponderale di circa 13 kg). Si è quindi deciso di eseguire un nuovo studio coronarografico con la conferma di coronarie esenti la lesioni ateromasiche significative.
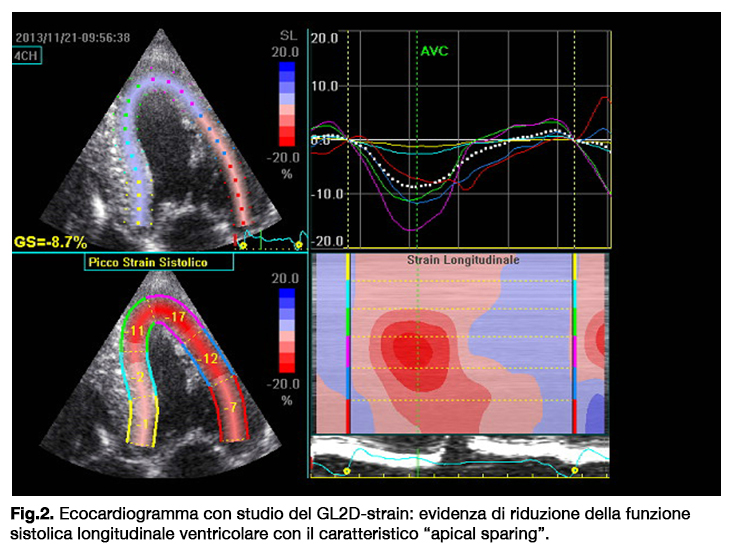
Si è quindi deciso di ripetere l’ecocardiogramma completandolo con lo studio del global longitudinal strain (GL2D-strain) con il riscontro di una severa riduzione della funzione sistolica longitudinale ventricolare (Global 2D strain = – 8.7%) con ‘risparmio delle porzioni apicali’ (rapporto apice/segmenti basale e medio = 1.5) (Fig.2)
Il paziente è stato quindi valutato dal collega nefrologo che, in considerazione del quadro ecocardiografico sospetto per cardiomiopatia infiltrativa e della presenza di alcuni elementi a favore di possibile Amiloidosi di tipo AL (epatomegalia con aumento degli indici di colestasi, ipotensione), ha consigliato di eseguire elettroforesi proteica urinaria, immunofissazione siero-urine, dosaggio catene leggere libere nel siero e prelievo del grasso periombelicale. Tali accertamenti sono tuttavia risultati tutti negativi.
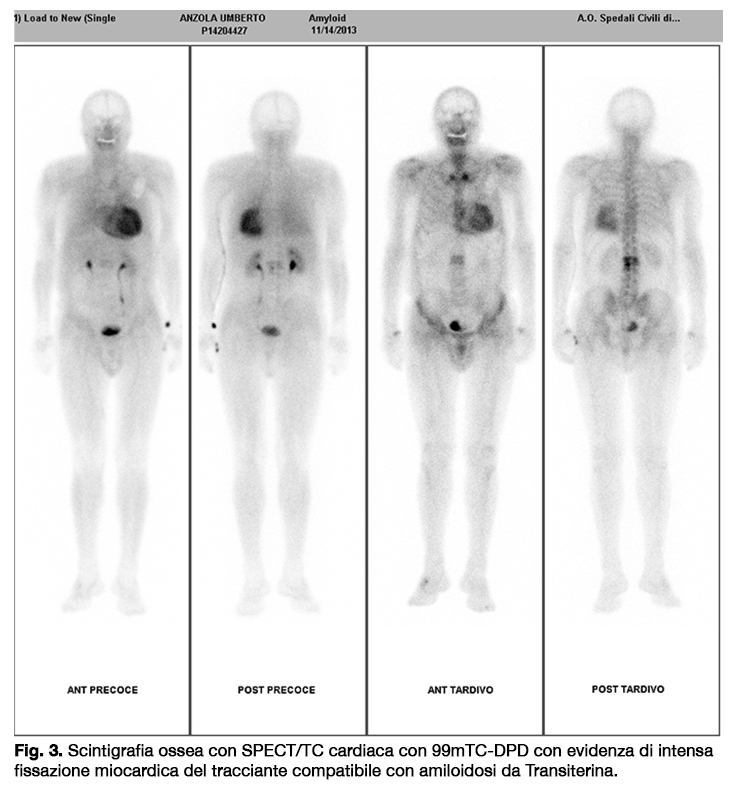
In considerazione dei pregressi interventi per sindrome del tunnel carpale bilaterale e del quadro elettroneurografico ed elettromiografico indicativi per neuropatia periferica sensitivo-motoria diffusa, è stato posto il sospetto di Amiloidosi da transtiretina. Il paziente è stato pertanto sottoposto a Scintigrafia ossea con SPECT/TC cardiaca con 99mTC-DPD che ha evidenziato intensa fissazione miocardica del tracciante, reperto compatibile con tale ipotesi diagnostica (Fig. 3).
Il paziente è stato pertanto inviato presso il Centro per lo Studio delle Amiloidosi sistemiche di Pavia per ulteriori approfondimenti diagnostico-terapeutici. La diagnosi genetica effettuata presso l’Istituto San Matteo di Pavia ha confermato la presenza della mutazione Ile68Leu in eterozigosi. È stato inoltre iniziato trattamento con Tafamidis.
Discussione
Il termine Amiloidosi fa riferimento ad un gruppo eterogeneo di patologie acquisite o ereditarie, localizzate o sistemiche, che condividono la caratteristica deposizione extracellulare di proteine fibrillari insolubili, con conseguente disorganizzazione dell’architettura tissutale degli organi coinvolti (1-2). La cardiomiopatia amiloidotica è una cardiomiopatia restrittiva associata ad incremento degli spessori parietali ed aumentata rigidità strutturale che causa rapido incremento delle pressioni intraventricolari in associazione solo ad un lieve incremento dei volumi di riempimento (3-4). Oltre al più evidente coinvolgimento parietale (responsabile della principale manifestazione clinica della cardiomiopatia, lo scompenso cardiaco), l’amiloide infiltra i vasi intramiocardici (determinando ischemia miocardica), gli atri (favorendo la comparsa di fibrillazione atriale) ed il sistema di conduzione determinando la comparsa di blocchi di branca ed occasionalmente di blocchi seno-atriali ed atrio-ventricolari talora con necessità di impianto di pace-maker. Il coinvolgimento cardiaco in corso di Amiloidosi può essere riconducibile a tre principali forme eziologiche distinte: l’Amiloidosi di tipo AL (secondaria alla presenza di cloni plasmacellulari nel midollo osseo che producono catene libere circolanti delle immunoglobuline) e le due forme (ereditaria e wild-type) di Amiloidosi transtiretino-relate (5).
La transtiretina (TTR) umana è una proteina che viene in larga parte prodotta dal fegato e circola nel sangue legata alla tiroxina e al complesso retinolo/vitamina A. La struttura nativa è formata da un omotetramero formato 4 sub-unità identiche legate tra loro in maniera covalente. Nei soggetti affetti da Amiloidosi, per motivi ancora in gran parte sconosciuti, la proteina ‘decide di disaggregarsi” formando dapprima dei monomeri e poi delle fibrille che tendono a depositarsi nei vari tessuti.
La Amiloidosi transtiretino relata familiare (ATTR) è la forma di Amiloidosi ereditaria più frequente e la prima ad essere stata identificata. Sono malattie a trasmissione autosomica dominante in cui un solo allele mutato è necessario per sviluppare la patologi, pertanto la maggior parte degli individui affetti è eterozigote per la mutazione patogena e produce sia la TTR normale che la variante mutata. Il gene che codifica per la TTR è collocato sul cromosoma 18 e nel corso delle ultime decadi sono state identificate più di 120 mutazioni di questo gene da cui derivano altrettante varianti proteiche. La maggior parte di queste mutazioni il più delle volte sono il risultato della sostituzione di un singolo nucleotide nel gene TTR; alcune mutazioni sono significativamente ricorrenti in alcune regioni geografiche. Esiste una precisa correlazione genotipo-fenotipo, anche se i fenotipi sono essenzialmente tre: forme con prevalente interessamento cardiaco, forme a prevalente interessamento neurologico e forme miste (6).
Questo ampio spettro di presentazioni cliniche rende il riconoscimento di ATTR particolarmente difficile, soprattutto in ambito cardiologico, qualora il quadro sia caratterizzato da un interessamento cardiaco esclusivo o prevalente, in assenza di altri elementi caratteristici che possano indirizzare verso la corretta diagnosi poiché può simulare altre cardiomiopatie non amiloidotiche (in particolare la cardiomiopatia ipertrofica) (7).
L’ecocardiogramma rappresenta il principale strumento per una diagnosi non invasiva di cardiomiopatia amiloidotica. Esso è genericamente caratterizzato dall’ispessimento delle pareti del ventricolo sinistro in assenza di dilatazione ventricolare sinistra. Indizi aggiuntivi per una cardiomiopatia infiltrativa includono l’ispessimento della parete libera del ventricolo destro e del setto interatriale, una significativa dilatazione bi-atriale, il diffuso ispessimento delle valvole atrio-ventricolari (> 5 mm) ed un lieve versamento pericardico. La frazione d’eiezione spesso è normale ma sia la velocità del wall motion ventricolare sia lo strain/strain rate del miocardio ventricolare sono frequentemente depresse. In modo analogo quando l’accorciamento radiale è ancora conservato risultano già, e quindi precocemente, alterate la contrazione longitudinale e la torsion ventricolare sinistra. Tali dati si possono acquisire sia con la metodica TDI sia con lo speckle tracking imaging (8). L’analisi combinata del pattern transmitralico e del TDI indica elevati valori della pressione di riempimento telediastolica ventricolare sinistra e/o un pattern di riempimento restrittivo.
Una tecnica diagnostica utile per la diagnosi di cardiomiopatia amiloidotica è la risonanza magnetica cardiaca con gadolinio. In presenza di amiloide infatti il gadolinio presenta un alterata cinetica caratterizzata da un late enhancement miocardico con una distribuzione diffusa subendocardica.
Un ulteriore esame diagnostico a disposizione è rappresentato dalla Scintigrafia ossea con SPECT/TC cardiaca con 99mTC-DPD: questo tracciante identifica il deposito miocardico di amiloide nella forma TTR relata ma non in quella da AL facilitando la diagnosi differenziale tra l’Amiloidosi da transtiretina da quella AL nella pratica clinica (9). La captazione miocardica del tracciante è correlata in modo lineare con la massa ventricolare sinistra e rappresenta un determinante prognostico dell’outcome nella ATTR sia da solo che in combinazione con lo spessore parietale del ventricolo sinistro (10).
Una volta posta la diagnosi si Amiloidosi TTR-relata è essenziale distinguere tra la forma ereditaria e la forma wild-type mediante l’analisi genetica per la ricerca di eventuali mutazioni patogene del gene della TTR.
Poiché la causa fondamentale alla base dell’Amiloidosi è rappresentata dall’instabilità dei mono – oligomeri di TTR, una della possibili soluzioni terapeutiche che ad oggi sono state studiate risiede nell’utilizzo di molecole ‘stabilizzatrici’ del tetramero di TTR che legandosi a siti specifici della proteina ne impediscono la frammentazione in monomeri, ovvero i precursori dei depositi fibrillari. Tafamidis, approvato dall’EMA nel 2011 per il trattamento della polineuropatia sintomatica nei pazienti adulti con ATTR, è uno stabilizzatore orale della transtiretina. Nello studio clinico di fase III pubblicato poco più di un anno fa, l’utilizzo di Tafamidis ha dimostrato, in pazienti affetti da ATTR acquisita o ereditaria, una significativa riduzione della mortalità per tutte le cause e della frequenza di ospedalizzazioni per cause cardiovascolari rispetto a placebo (11). Sulla base di questi risultati il comitato per i medicinali per uso umano dell’EMA nel Dicembre del 2019 ha raccomandato l’approvazione di Tafamidis anche per il trattamento delle forme di Cardiopatia amiloidotica transtiretino-relate.
Bibliografia
- Falk RH, Dubrey S.W. Amyloid Heart disease. Prog Cardiovasc Dis 2010;52:347–361.
- Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart: a comprehensive review. Arch Intern Med 2006;166:1805–1813.
- Maron BJ, Towbin JA, Thiene G, et al. Contemporary Definitions and Classification of the Cardiomyopathies: An American Heart Association Scientific Statement From the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006;113:1807–1816
- Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. 2008;29:270–276.
- Rapezzi C, Quarta CC, Riva L, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol.2010 Jul;7(7):398-408.
- Maurer MS, Hanna M, Grogan M, et al. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016 Jul 12;68(2):161-72. doi: 10.1016/j.jacc.2016.03.596.
- Selvanayagam JB, Hawkins PN, Paul B, et al.. Evaluation and management of the cardiac amyloidoses. J Am Coll Cardiol 2007;50:2101–10.
- Koyama J, Davidoff, R & Falk RH. Longitudinal myocardial velocity gradient derived from pulsed Doppler tissue imaging in AL amyloidosis: a sensitive indicator of systolic and diastolic dysfunction. J Am Soc Echocardiogr 2004;17:36–44.
- Rapezzi C, Quarta CC, Guidalotti PL, et al. Usefulness and limitations of 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid 88 scintigraphy in the aetiological diagnosis of amyloidotic cardiomyopathy. Eur J Nucl Med Mol Imaging 2011;38:470–8.
- Rapezzi C, Quarta CC, Guidalotti PL, et al. Role of (99m)Tc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging 2011;4:659–70.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018;379:1007-16